
NEM 2A ou syndrome de Sipple (mutations du gène RET)
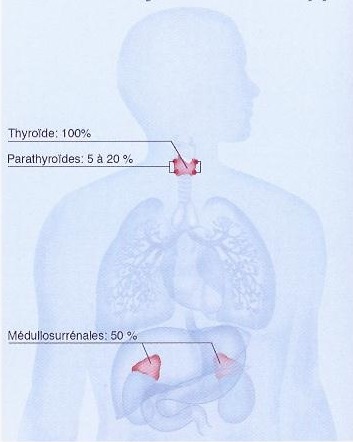
Le cancer médullaire de la thyroïde (CMT) est un cancer rare qui se développe aux dépens des cellules C parafolliculaires thyroïdiennes responsables de la sécrétion de calcitonine. Le CMT représente 5-10% des cancers de la thyroïde. Son incidence en pathologie nodulaire thyroïdienne se situe aux alentours de 1-2 %.
Sa prévalence dans la population générale est estimée à 1/14 300. Le CMT peut se révéler par un nodule thyroïdien avec euthyroïdie ou un goitre multinodulaire, associé le plus souvent à des adénopathies satellites. Il se présente sous deux formes : sporadique majoritaire et familiale dans près de 30 % des cas : il s'intègre alors dans la néoplasie endocrinienne multiple de type 2 (affection héréditaire monogénique rattachée à des mutations germinales du gène RET, marqueur génétique du CMT. En savoir +